6. Atoms¶
6.1. Input crystal data¶
The attributes of the Atoms object can be populated in one of three
ways, by reading an atoms.inp file, by reading a CIF
file, or by setting the
attributes programmatically.
The format of the atoms.inp file is documented elsewhere.
Importing data from one is as simple as specifying the file when the
Atoms object is created. In this example, an atoms.inp file is
imported and an input file for FEFF6 is written to standard
output.
#!/usr/bin/perl
use Demeter;
my $atoms = Demeter::Atoms->new(file => "ybco.inp");
print $atoms->Write("feff6");
The command-line ATOMS program that comes with DEMETER is longer than 5 lines, but does not really do much beyond this example.
Importing crystal data from a CIF file is no more difficult, however
you need to explicitly tell the Atoms object that the input data is in
CIF format. The Atoms object assumes that the value of the file
attribute points at an atoms.inp file, so the cif
attribute must be used to specify a CIF file.
#!/usr/bin/perl
use Demeter;
my $atoms = Demeter::Atoms->new(cif => "Fe2N_ICSD.cif");
print $atoms->Write("feff6");
The STAR::Parser module is used to interpret the CIF file. This is a quirky bit of code and my understanding of it is not so deep. There are probably examples of CIF files that do not get imported properly, but it seems to work in most cases. You should consider a valid CIF file that cannot be imported by DEMETER to be a reportable DEMETER bug.
CIF files can contain more than one structure. By default,
DEMETER will import the first structure contained in the
CIF file. To specify another structure, use the record
attribute. Note that the counting is zero based, so this example is
importing the second record in the CIF file. In a GUI like
ARTEMIS, it is probably wise to let the user count from 1
and to do the translation behind the scenes.
#!/usr/bin/perl
use Demeter;
my $atoms = Demeter::Atoms->new(cif => "AuCl.cif", record=>1);
print $atoms->Write("feff6");
The Artemis document
offers more discussion of the content of the atoms.inp file.
6.2. Object methods¶
6.2.1. Manually inputing crystal data¶
Starting with this atoms.inp file
title YBCO: Y Ba2 Cu3 O7
space P M M M
rmax=5.2 a 3.823 b 3.886 c 11.681
core=cu2
atom
! At.type x y z tag
Y 0.5 0.5 0.5
Ba 0.5 0.5 0.184
Cu 0 0 0 cu1
Cu 0 0 0.356 cu2
O 0 0.5 0 o1
O 0 0 0.158 o2
O 0 0.5 0.379 o3
O 0.5 0 0.377 o4
you can manually load up the attributes of the Atoms object. This is what the ATOMS interface in ARTEMIS does. A straight-forward, brute-force approach is shown in this example:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 | #!/usr/bin/perl
use Demeter;
my $atoms = Demeter::Atoms->new();
$atoms -> set(a=>3.823, b=>3.886, c=>11.681);
$atoms -> space('P M M M');
## add each site
$atoms -> push_sites( join("|", 'Y', 0.5, 0.5, 0.5, 'y' ) );
$atoms -> push_sites( join("|", 'Ba', 0.5, 0.5, 0.184, 'ba' ) );
$atoms -> push_sites( join("|", 'Cu', 0.0, 0.0, 0.0, 'cu1') );
$atoms -> push_sites( join("|", 'Cu', 0.0, 0.0, 0.356, 'cu2') );
$atoms -> push_sites( join("|", 'O', 0.0, 0.5, 0.0, 'o1' ) );
$atoms -> push_sites( join("|", 'O', 0.0, 0.0, 0.158, 'o2' ) );
$atoms -> push_sites( join("|", 'O', 0.0, 0.5, 0.379, 'o3' ) );
$atoms -> push_sites( join("|", 'O', 0.5, 0.0, 0.377, 'o4' ) );
$atoms -> core('cu2');
$atoms -> set(rpath=>5.2, rmax => 8);
print $atoms->Write("feff6");
|
Once all the data is set, simply call the Write method and the
object will take care of populating the cell and explanding the cluster.
Note the odd syntax in lines 8 through 15 for loading the sites
attribute. The elements of that array are strings of
vertical-bar-separated (awkward, perhaps, but that's how it works)
values of
- element symbol
- fractional x coordinate
- fractional y coordinate
- fractional z coordinate, and
- tag.
Note that the tag has a limit of 10 characters.
At line 16, the central atom is chosen by specifying a valid tag as the
value of the core attribute.
6.3. Other methods¶
Todo
- Absorption calculations: xsec, deltamu density mcmaster i0 selfsig selfamp netsig
- Mentions cluster and nclus attributes
6.4. Output¶
Output from the Atoms object is handled by the Write method. Note
that this is capitalized to avoid any possible confusion (by perl or by
a syntax highlighting text editor) with perl's write
function, as shown in
this example:
#!/usr/bin/perl
use Demeter;
my $atoms = Demeter::Atoms->new(file => "ybco.inp");
print $atoms->Write("feff6");
There are several output targets, which are formatted using
templates from the Atoms template set.
The output targets, i.e. the arguments of the Write method, that
come with DEMETER are:
feff6- Input file for FEFF6.
feff7- Input file for FEFF7 (which is not really very different from the FEFF6 input file).
feff8- Input file for FEFF8.
atoms- Input file for ATOMS. This used as the save-file target for a GUI.
p1Input file for ATOMS using the
P 1spacegroup and the fully populated unit cell. Here's an example:title = YBCO: Y Ba2 Cu3 O7 space = P M M M a = 3.82300 b = 3.88600 c = 11.68100 alpha = 90.00000 beta = 90.00000 gamma = 90.00000 rmax = 5.20000 core = cu2 shift = atoms # el. x y z tag Y 0.50000 0.50000 0.50000 Y Ba 0.50000 0.50000 0.18400 Ba Ba 0.50000 0.50000 0.81600 Ba Cu 0.00000 0.00000 0.00000 cu1 Cu 0.00000 0.00000 0.35600 cu2 Cu 0.00000 0.00000 0.64400 cu2 O 0.00000 0.50000 0.00000 o1 O 0.00000 0.00000 0.15800 o2 O 0.00000 0.00000 0.84200 o2 O 0.00000 0.50000 0.37900 o3 O 0.00000 0.50000 0.62100 o3 O 0.50000 0.00000 0.37700 o4 O 0.50000 0.00000 0.62300 o4
absorptionA file containing several interesting calculations using tables of absorption coefficients. Here's an example:
## --*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*-- ## total mu*x=1: 8.160 microns, unit edge step: 23.243 microns ## specific gravity: 6.375 ## --*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*-- ## normalization correction: 0.00046 ang^2 ## --*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--*--
spacegroupA file containing a description of the space group. Here's an example:
# title = YBCO: Y Ba2 Cu3 O7 # space = P M M M # a = 3.82300 b = 3.88600 c = 11.68100 # alpha = 90.00000 beta = 90.00000 gamma = 90.00000 # rmax = 5.20000 core = cu2 # shift = # atoms # # el. x y z tag # Y 0.50000 0.50000 0.50000 Y # Ba 0.50000 0.50000 0.18400 Ba # Cu 0.00000 0.00000 0.00000 cu1 # Cu 0.00000 0.00000 0.35600 cu2 # O 0.00000 0.50000 0.00000 o1 # O 0.00000 0.00000 0.15800 o2 # O 0.00000 0.50000 0.37900 o3 # O 0.50000 0.00000 0.37700 o4 Spacegroup P M M M (#47) Schoenflies: D_2h^1 Full symbol: p 2/m 2/m 2/m New symbol : Thirtyfive : Nicknames : Common shift vector: Bravais translations: 8 positions: x y z -x -y z -x y -z x -y -z -x -y -z x y -z x -y z -x y z
xyzandalchemy- These are two common formats for molecule visualization software.
overfullThis is an
xyzfile containing the contents of the fully decorated unit cell, also including atoms that are near to all the corners, edges, and walls. This helps to make a figure like this of a well-decorated unit cell: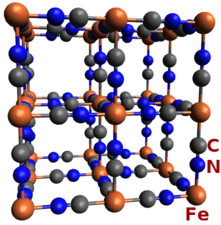
The ♦Atoms→overfull_margin parameter is used to set what is meant by “near” the remaining corners, edges, and walls.
DEMETER is copyright © 2009-2016 Bruce Ravel – This document is copyright © 2016 Bruce Ravel
This document is licensed under The Creative Commons Attribution-ShareAlike License.
If DEMETER and this document are useful to you, please consider supporting The Creative Commons.
