13.1. Example 1: FeS2¶
Iron pyrite, FeS2, is my favorite teaching example which I use to introduce people to fitting with ARTEMIS. It is a crystal with a well-known structure. I have beautiful data. And I am able to create a very successful fitting model. As a cubic crystal, the structure is not too complicated. However, there is some internal structure in the unit cell, making this a much less trivial example than, say, an FCC metal.
While this example certainly does not represent a typical research problem, it is an excellent introduction to the mechanics of using ARTEMIS and it results in a very satisfying fit.
You can find example EXAFS data and crystallographic input data at my XAS Education site. Import the μ(E) data into ATHENA. When you are content with the processing of the data, save an ATHENA project file and dive into this example.
13.1.1. Import data¶
After starting ARTEMIS,  click on the
Add button at the top of the Data sets
list in the Main window. This will open a file selection dialog.
Click to find the ATHENA project file containing the data
you want to analyze. Opening that project file displays the project
selection dialog.
click on the
Add button at the top of the Data sets
list in the Main window. This will open a file selection dialog.
Click to find the ATHENA project file containing the data
you want to analyze. Opening that project file displays the project
selection dialog.
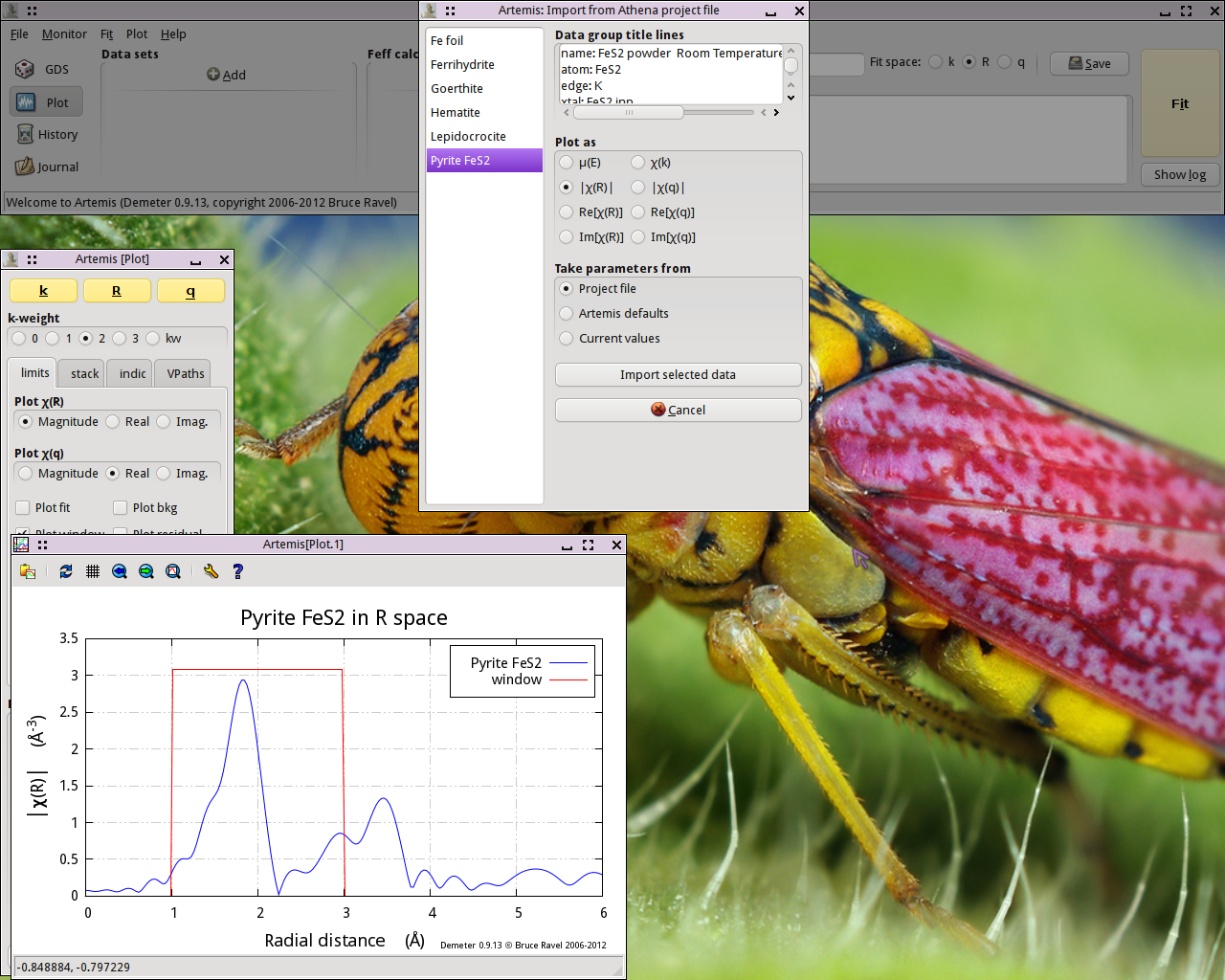
Fig. 13.1 Import data into ARTEMIS
The project file used here has several iron standards. Select FeS2 from the list. That data set gets plotted when selected.
Now click the Import button. That
data set gets imported into ARTEMIS. An entry for the FeS2 is created in the Data list, a window for interacting with
the FeS2 data is created, and the FeS2 data are
plotted as χ(k).
The next step is to prepare for the FEFF calculation using
the known FeS2 crystal structure.
Clicking on the line in the Data window that says Import
crystal data or a Feff calculation will post a file selection dialog.
Click to find the atoms.inp file containing the FeS2
crystal structure.
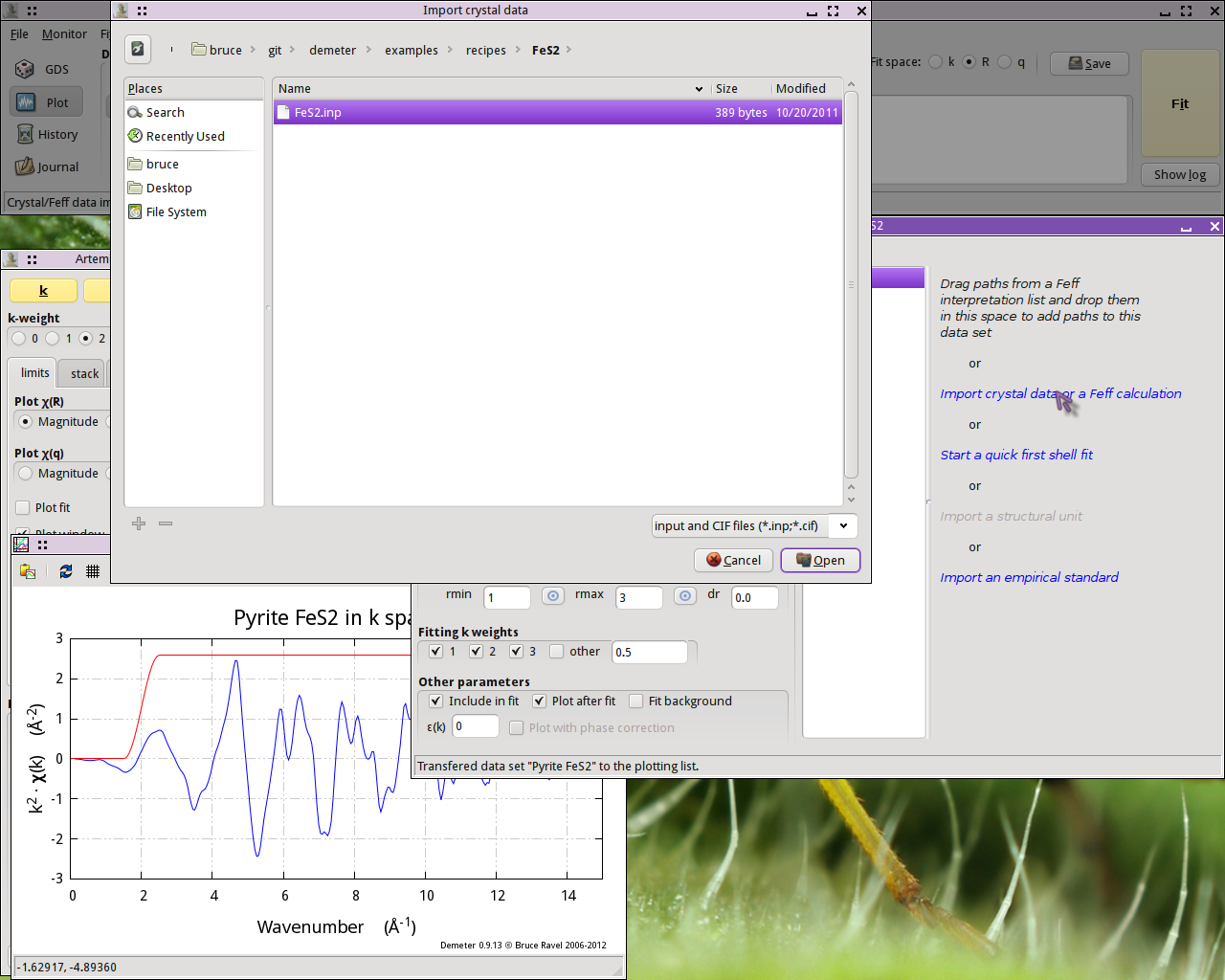
Fig. 13.2 Import crystal data into ARTEMIS
With the FeS2 crystal data imported, run ATOMS by
clicking the Run Atoms button on
the ATOMS tab of the FEFF windows. That will
display the FEFF tab containing the FEFF input
data. Click the Run Feff button
to compute the scattering potentials and to run the pathfinder.
Once the FEFF calculation is finished, the path
intepretation list is shown in the Paths tab. This is the list of
scattering paths, sorted by increasing path length. Select the first
11 paths by clicking on the path
0000, then Shift- clicking
on path 0010. The selected paths will be highlighted.
Click on one of the highlighted paths and,
without letting go of the mouse button,  drag the paths
over to the Data window and drop them onto the empty Path list.
drag the paths
over to the Data window and drop them onto the empty Path list.
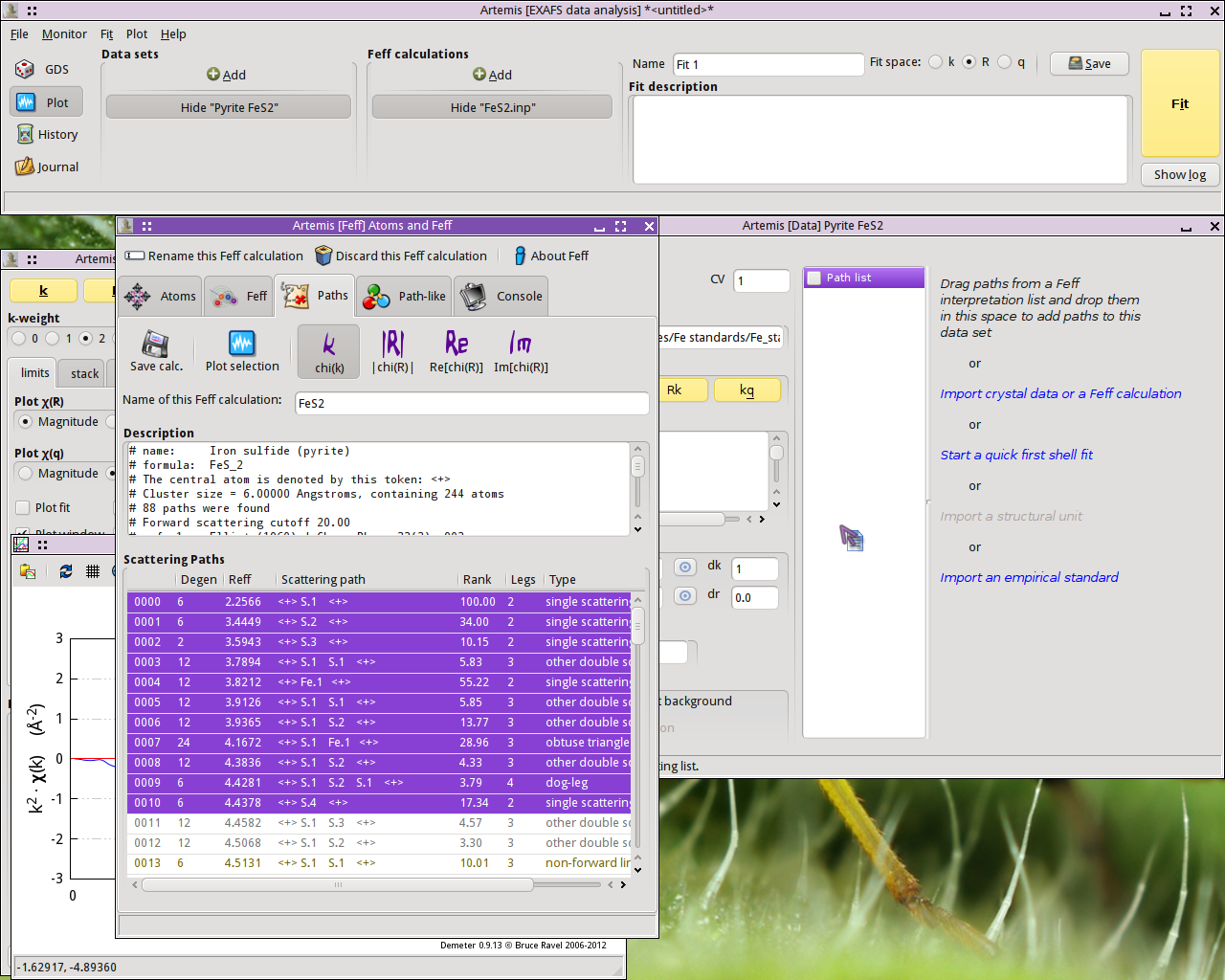
Fig. 13.3 Drag and drop paths onto a data set
Dropping the paths on the Path list will associate those
paths with that data set. That is, that group of paths is now
available to be used in the fitting model for understanding the FeS2 data.
Each path will get its own Path page. The Path page for a path is displayed when that path is clicked upon in the Path list. Shown below is the FeS2 data with its 11 paths. The first path in the list, the one representing the contribution to the EXAFS from the S single scattering path at 2.257 Å, is currently displayed.
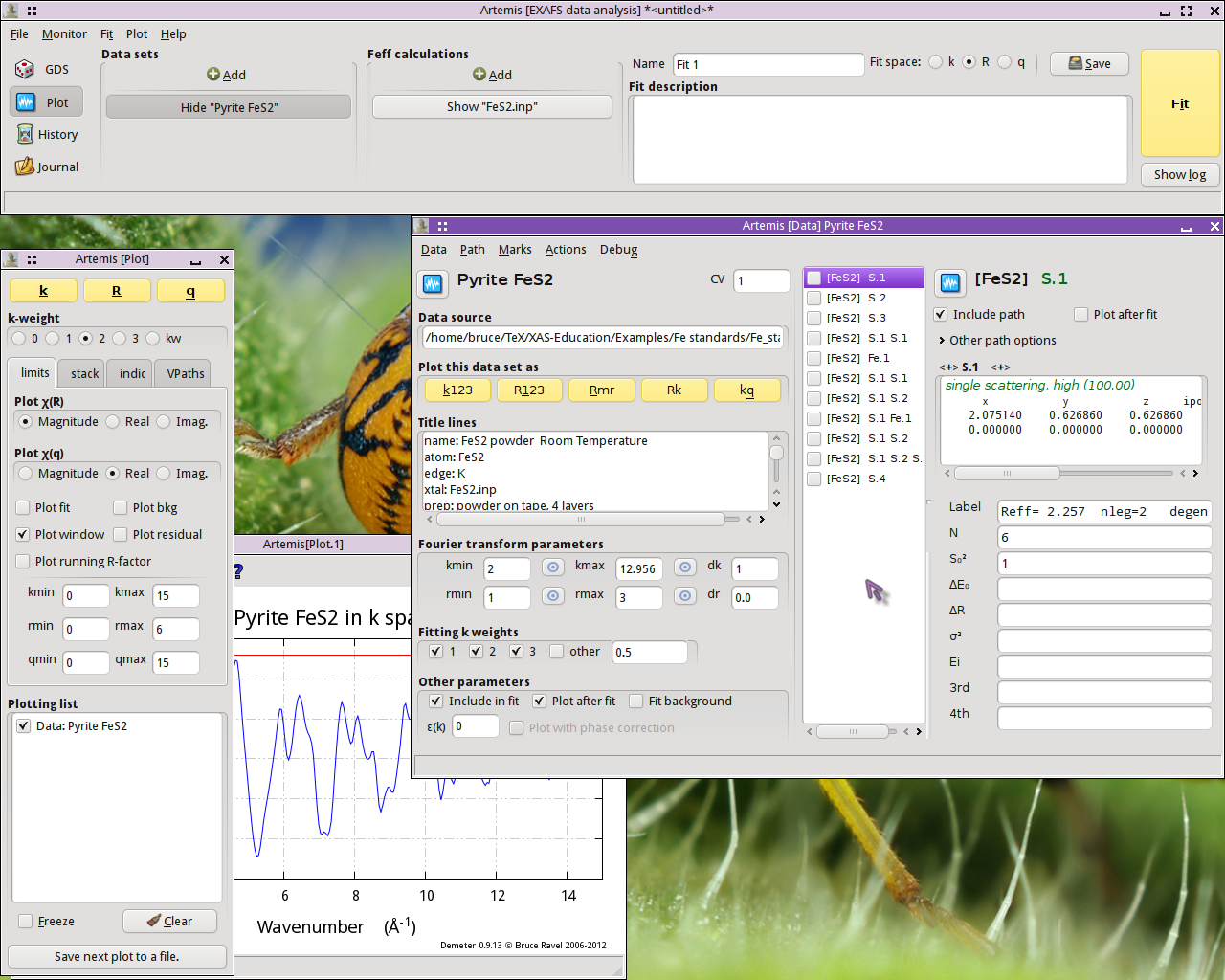
Fig. 13.4 Paths associated with a data set
13.1.2. Examine the scattering paths¶
The first chore is to understand how the various paths from the FEFF calculation relate to the data. To this end, we need to populate the Plotting list with data and paths and make some plots.
First let's examine how the single scattering paths relate to the
data. Mark each of the first four single scattring paths – the
ones labeled S.1, S.2, S.3, and
Fe.1 – by clicking on their check
buttons. Transfer those four paths to the Plotting list by selecting
.
With the Plotting list poluated as shown below,
click on the R plot button in the Plot window to make
the plot shown.
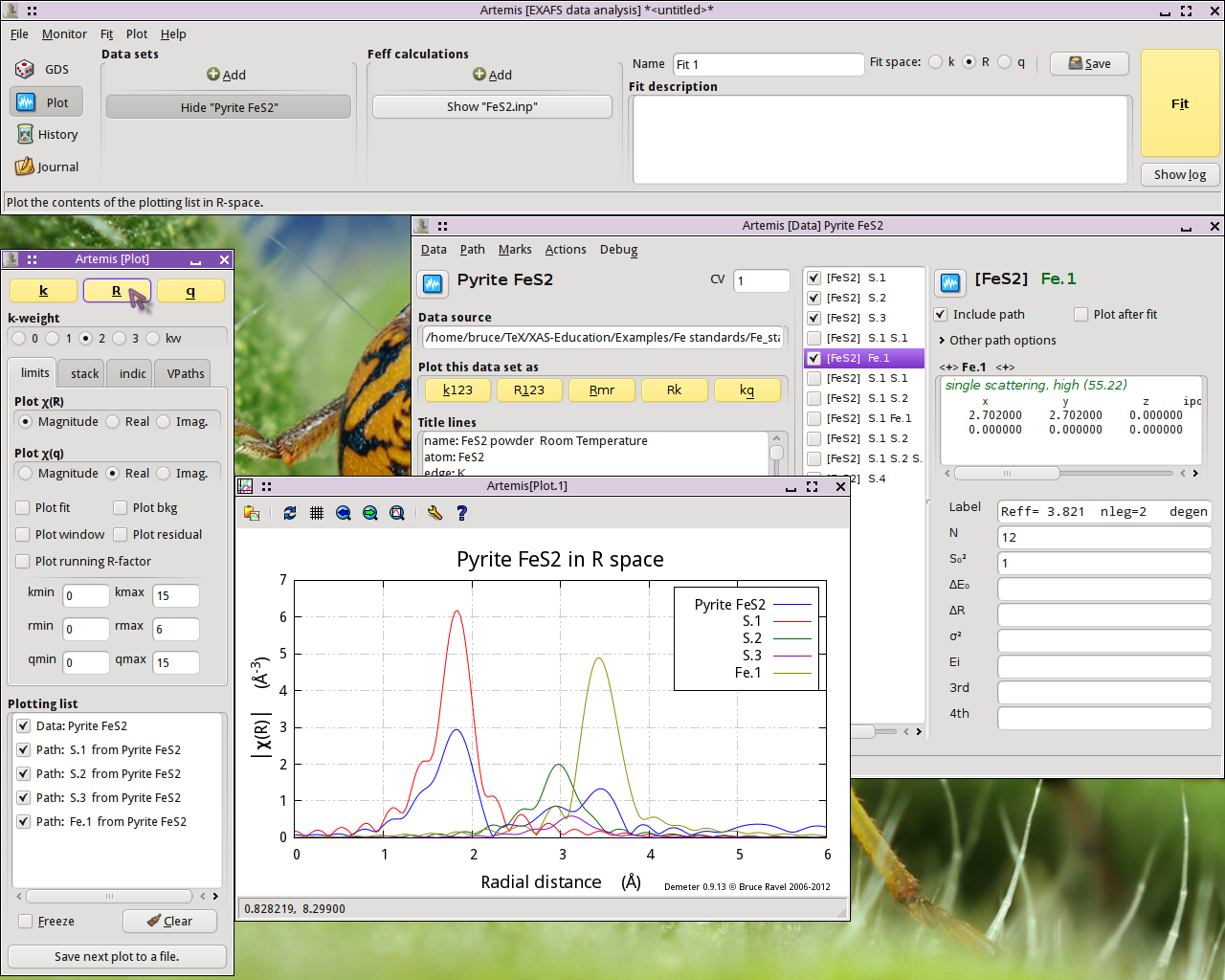
Fig. 13.5 FeS2 data plotted with the first four single scattering paths
The first interesting thing to note is that the first peak in the data seems to be entirely explained by the path from the S atom at 2.257 Å. None of the other single scattering paths contribute significantly to the region of R-space.
The second interesting thing to note is that the next three single scattering paths are not so well separated from one another. While it may be tempting to point at the peaks at 2.93 Å and 3.45 Å and assert that they are due to the second shell S and the fourth shell Fe, it is already clear that the situation is more complicated. Those three single scattering paths overlap one another. Each contriobutes at least some spectral weight to both of the peaks at 2.93 Å and 3.45 Å.
The first peak shold be reather simple to interpret, but higher shells are some kind of superposition of many paths.
What about the multiple scattering paths?
To examine those, first clear the Plotting list by
clicking the Clear button at the
bottom of the Plot window. Transfer the FeS2 data back to the
Plotting list by clicking its transfer button:
 . Mark the first three multiple scattering paths by
clicking their mark buttons. Select
.
. Mark the first three multiple scattering paths by
clicking their mark buttons. Select
.
With the Plotting list newly populated, make a new plot of | χ(R)|.
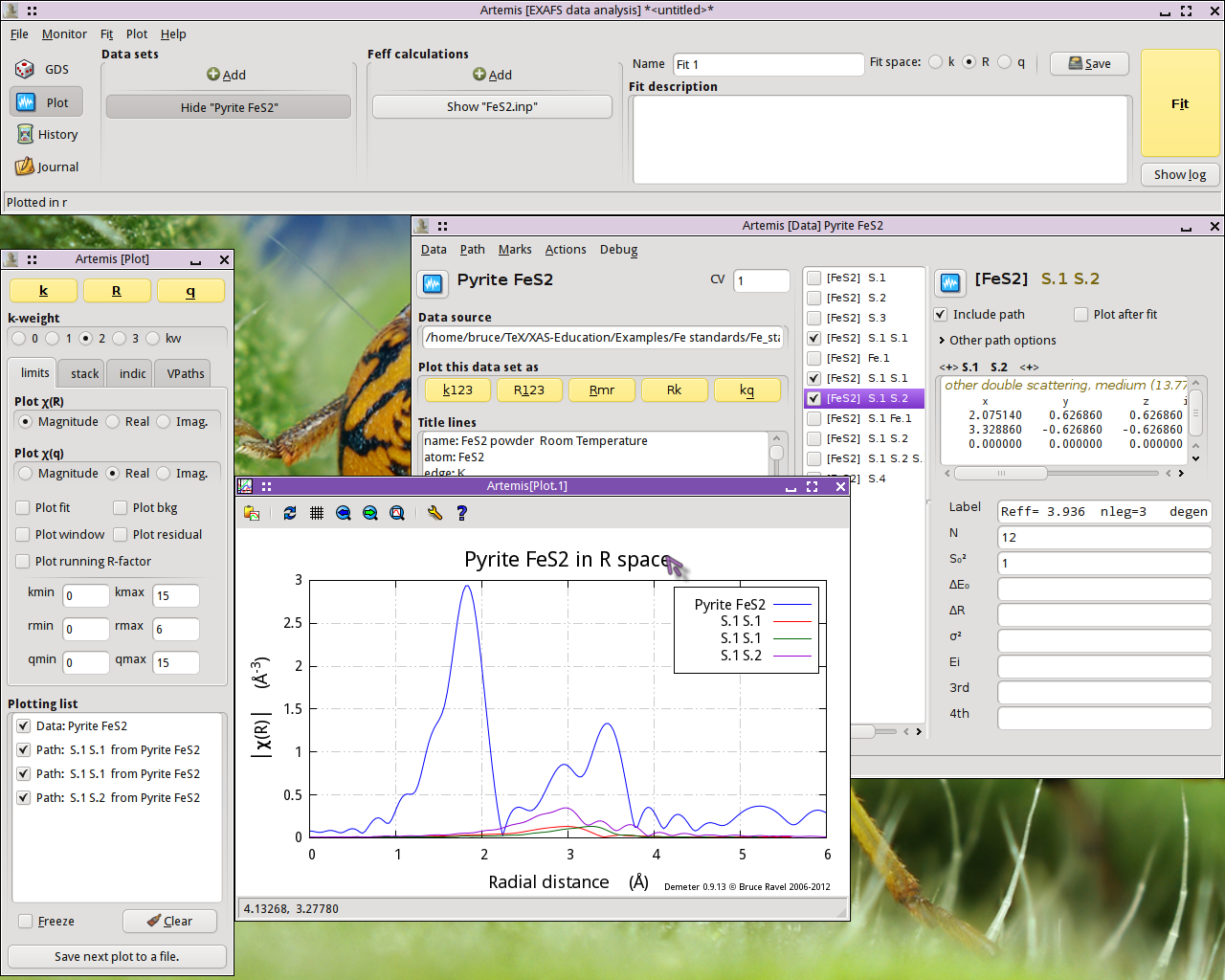
Fig. 13.6 FeS2 data plotted with the first three multiple scattering paths
The two paths labeled S.1 S.1, which represent two different ways for the photoelectron to scatter from a S atom in the first coordination shell then scatter from another S atom in the first coordination shell, contribute rather little spectra weight. Given their small size, it seems possible that we may be able to ignore those paths when we analyze our FeS2 data.
The S.1 S.2 path, which first scatters from a S in the first coordination shell then from a S in the second coordination shell, contributes significantly to the peak at 2.93 Å. It seems unlikely that we will be able to ignore that path.
To examine the next three multiple scattering paths, clear the Plotting list, mark those paths, and repopulate the Plotting list.
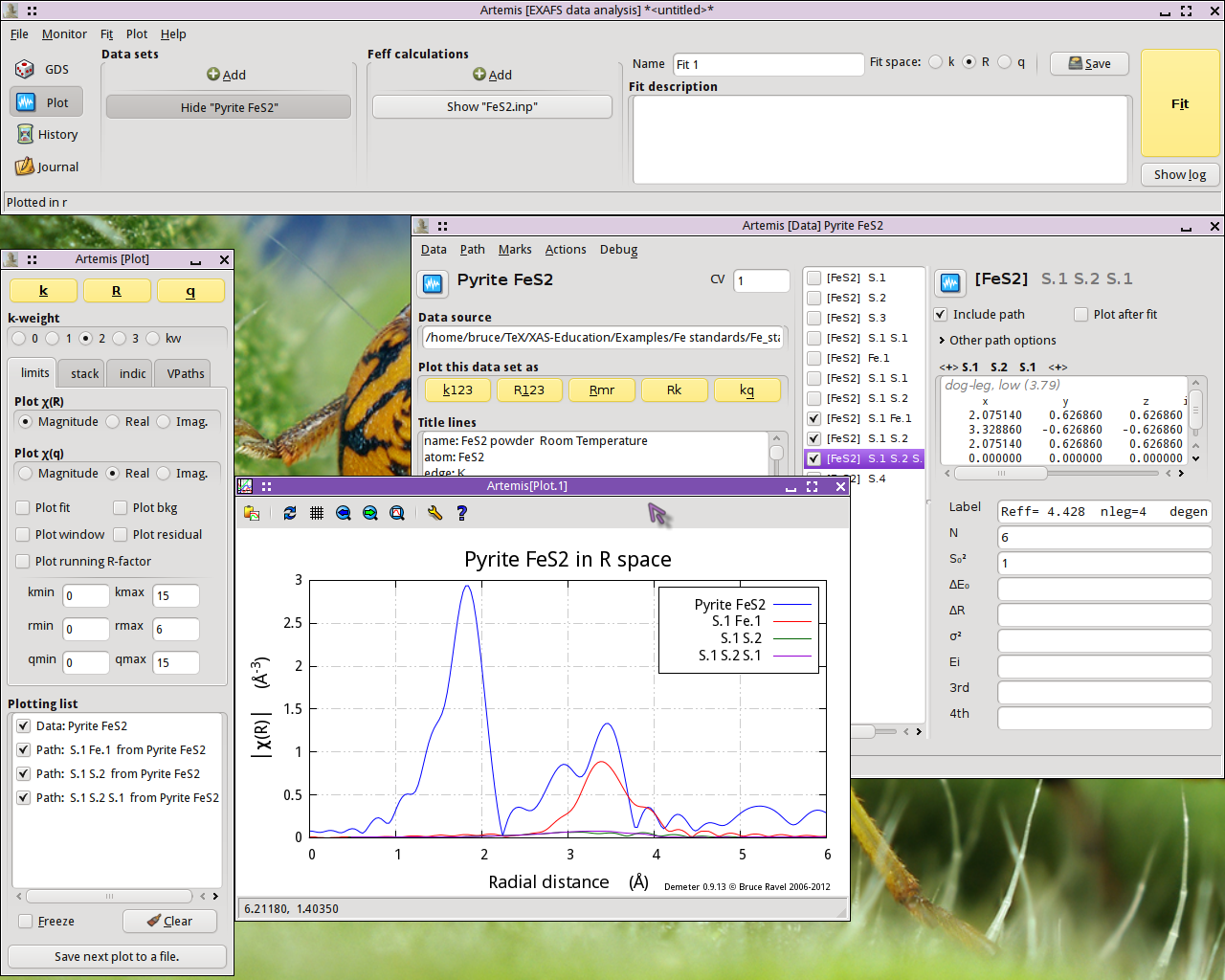
Fig. 13.7 FeS2 data plotted with the next three multiple scattering paths
The S.1 Fe.1 path, which scatters from a S atom in the first coordination shell then scatters from an Fe atom in the fourth coordination shell, is quite substantial. It will certainly need to be considered in our fit. The other two paths are tiny.
13.1.3. Fit to the first coordination shell¶
We begin by doing an analysis of the first shell. As we saw above, we only need the first path in the path list. To prepare for the fit, we do the following:
- Exclude all but the first path from the fit. With the first path selected in the path list and displayed, select . This will mark all paths except for the first one. Then select . This will exclude those paths from the fit. That is indicated by the triple parentheses in the path list.
- Set the values of Rmin and Rmax to cover just the first peak.
- For this simple first shell fit, we set up a simple, four-parameter
model. The parameters
amp,enot,delr, andssare defined in the GDS window and given sensible initial guess values. - The path parameters for the first shell path are set. S20 is set to
amp, E0 is set toenot, ΔR is set todelr, and σ2 is set toss.
Note that the current settings for k- and R-range result in a bit more than 7 independent points, as approximated using the Nyquist criterion. With only 4 guess parameters, this should be a reasonable fitting model.

Fig. 13.8 Setting up for a first shell fit
Now hit the Fit button. Upon completion of the fit, the following things happen:
- An “Rmr” plot is made of the data and the fit.
- The log Window is displayed with the results of the fit
- The Fit and plot buttons are recolored according to the evaluation of the happiness parameter.
- The Plotting list is cleared and repopulated with the data.
- The fit is entered into the History window (which is not in the screenshot below).
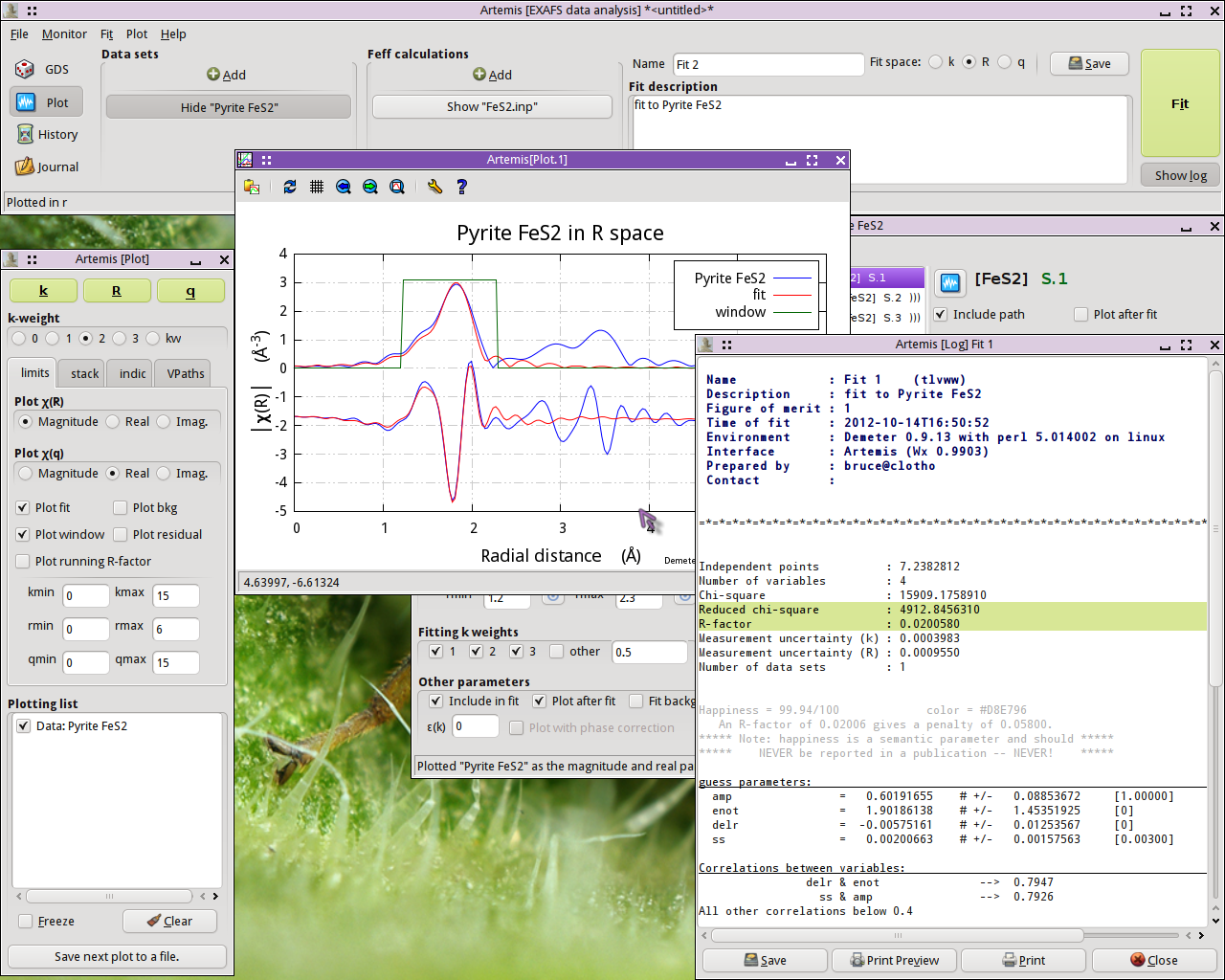
Fig. 13.9 Results of the first shell fit
This is not a bad result. The value of enot is small, indicating
that a reasonable value of E0 was chosen back in
ATHENA. delr is small and consistent with 0, as we
should expect for a known crystal. ss is a reasonable value with a
reasonable error bar. The only confusing parameter is amp, which
is a bit smaller than we might expect for a S20
value.
The correlations between parameters are of a size that we expect. The R-factor evaluates to about 2% misfit. χ2v is really huge, but that likely means that ε was not evaluated correctly. All in all, this is a reasonable fit.
13.1.4. Extending the fit to higher shells¶
Although we know that we will need to include some multiple scattering paths in this fit, we'll start by including the next three single scattering paths, adjusting the fitting model accordingly.
This fit will include the peaks in the range of 3 Å to 3.5 Å, so
set Rmax to 3.8. Next click on each of
next three single scattering paths – the ones labeled
S.2, S.3, and Fe.1, then
click on the Include path button for
each path.
At this point we need to begin considering the details of our fitting model more carefully. S20 and E0 should be the same for each path, so we will use the same guess parameters for each path.
This is a cubic material. Ignoring the possibility that fractional
coordinate of the S atom in the original crystal data might be a
variable in the fit and considering only the prospect of a volumetric
lattice expansion, we define
a guess parameter alpha and set the ΔR for each
path to alpha*reff.
Finally, we set independent σ2 parameters for each single scattering path. This gives a total of 7 guess parameters, compared to the Nyquist evaluation of about 16.5, given the new value of Rmax.
Things now look like this:
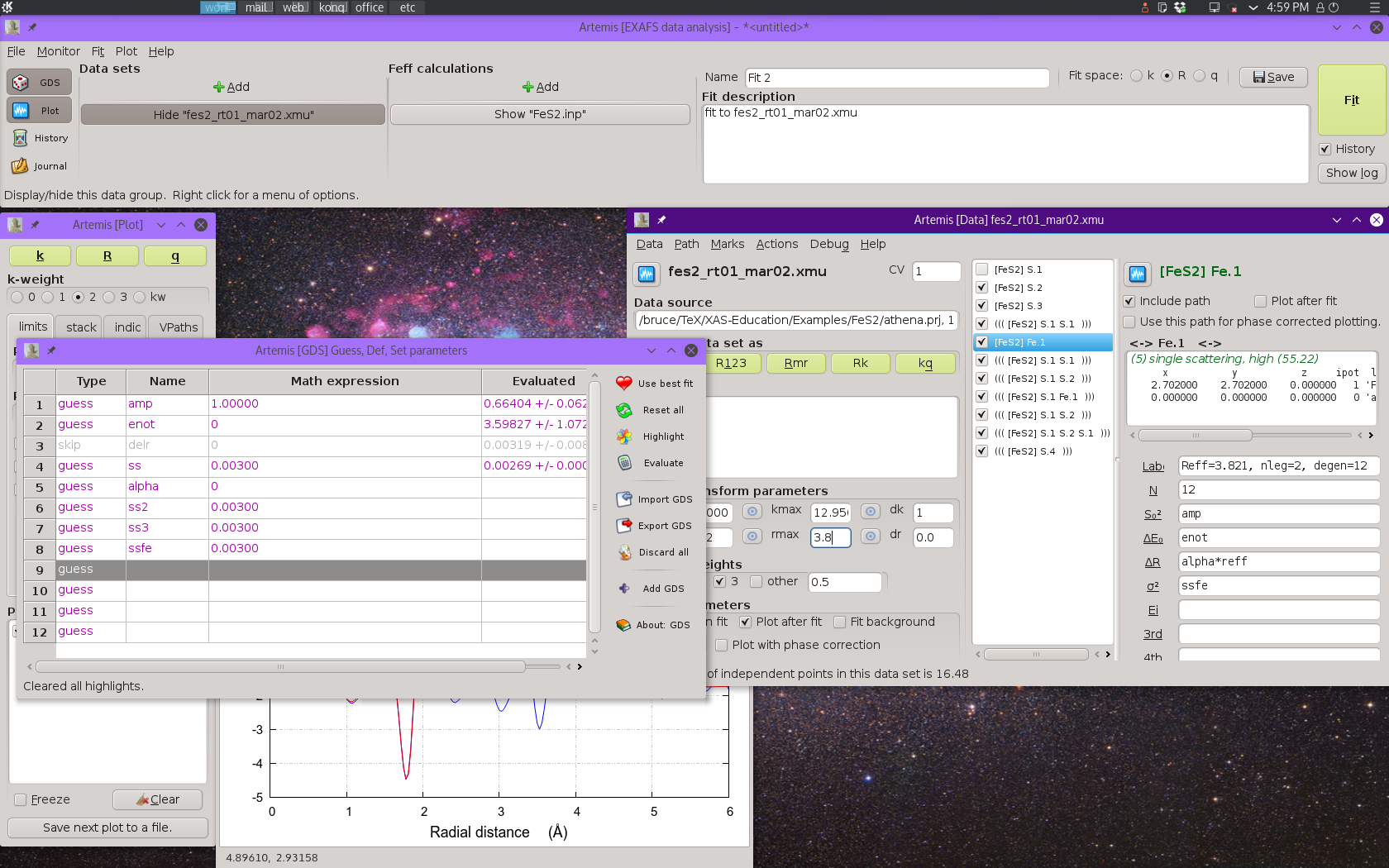
Fig. 13.10 Preparing to run the four-shell fit with only single scattering paths.
At this point we can run the fit and see how things go!
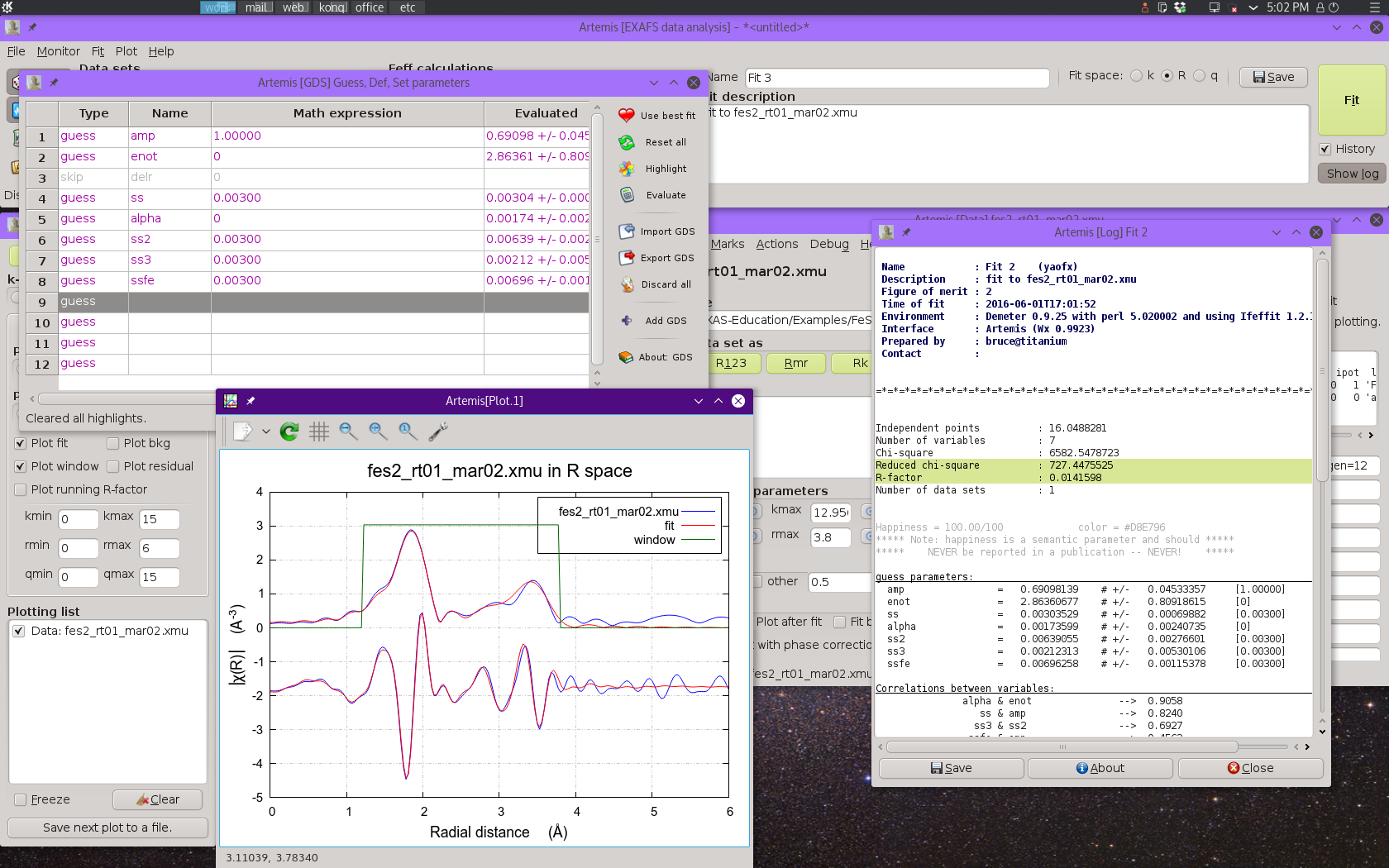
Fig. 13.11 Result of the four-shell fit using on the single scattering paths. Note the misfit around 3.2 Å.
This fit isn't bad. It does a good job of representing the data, with
the exception of some misfit around 3.2 Å. Most of the fitting
parameters are pretty reasonable, with the exception of the result for
ss3. This is discussed in some detail in the
lecture notes,
but the bottom line is that the signal from these two S scatterers,
while a significant contributor of Fourier components to the fit, is
not large enough to support its own robust σ2
parameter. We will make ss3 into a def parameters, setting
it equal to ss2.

Fig. 13.12 Making ss3 a def parameter.
At this point we need to introduce the two significant multiple
scattering paths. Start by clicking the
Include path button for the S.1 S.2 and
S.1 Fe.1 paths.
Make sure each of those paths has their S20, E0, and ΔR path parameters set to amp, enot, and
alpha*reff, respectively.
As discussed in the lecture notes,
we don't necessarily want to give these multiple scattering paths
independent σ2 parameters. Indeed, they are unlikely
to be robust fitting parameters for the same reason that ss3
proved not to be a robust parameter. Instead, we will approximate
these two σ2 parameters using guess parameters
already in the fit.
Consider these two triangular paths on a leg-by-leg basis. For the
S.1 Fe.1 path, the first and last leg are of the distance
between the absorber and the Fe scatterer in the fourth shell. The
remaining leg is the first shell distance. In terms of length, this
path covers the full distance of the fourth shell path and half the
distance of the first shell path. Thus we will approximate its
σ2 as ssfe+ss/2.
The S.1 S.2 triangle is a bit more ambiguous, since we do
not have a parameter describing the leg that goes from the first shell
S atom to the second shell S atom. In the absence of a better
approximation, we will use ss*1.5, i.e. something a bit bigger
than the first shell σ2. Neither of these are quite
right, but neither is ridiculously wrong. And parameterizing them in
this way allows us to avoid introducing a weak guess
parameter into the fitting model.
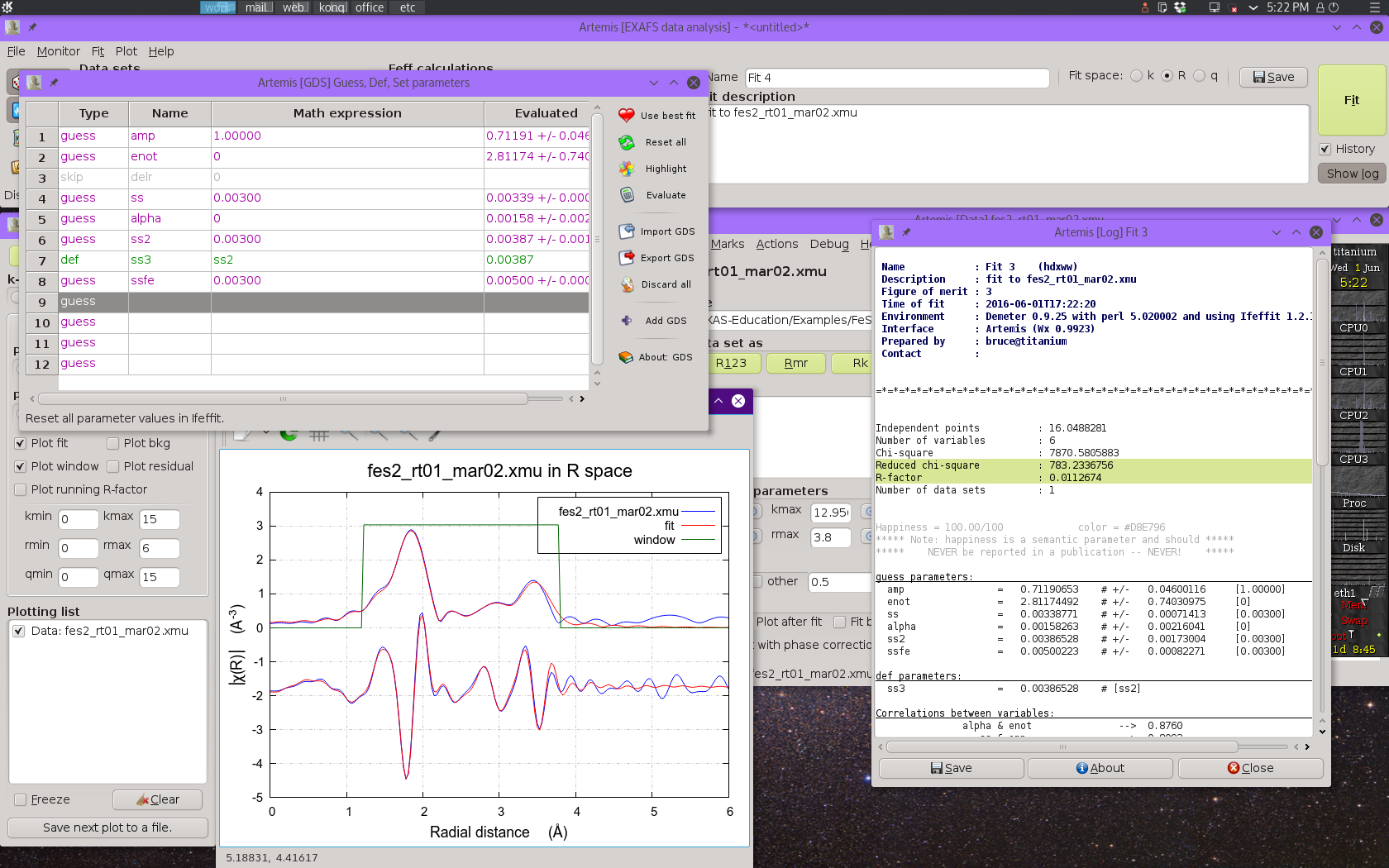
Fig. 13.13 Result of the four-shell fit including four single scattering paths and the two largest multiple scattering paths.
This fit does a much better job in the region around 3.2 Å, however the last bit of the fitting range is not quite right. The fitting parameters are all quite reasonable and the percentage misfit within the fitting range is quite small – around 1%.
13.1.5. The final fitting model¶
To clean up the last bit of the fitting range, we can include a set of three multiple scattering paths that represent collinear paths involving the first shell S atoms. An ARTEMIS project file for the final fitting model can be found among the examples at my XAS Education site. Adding these three paths and extending Rmax to 4.2 Å results in an excellent fit.
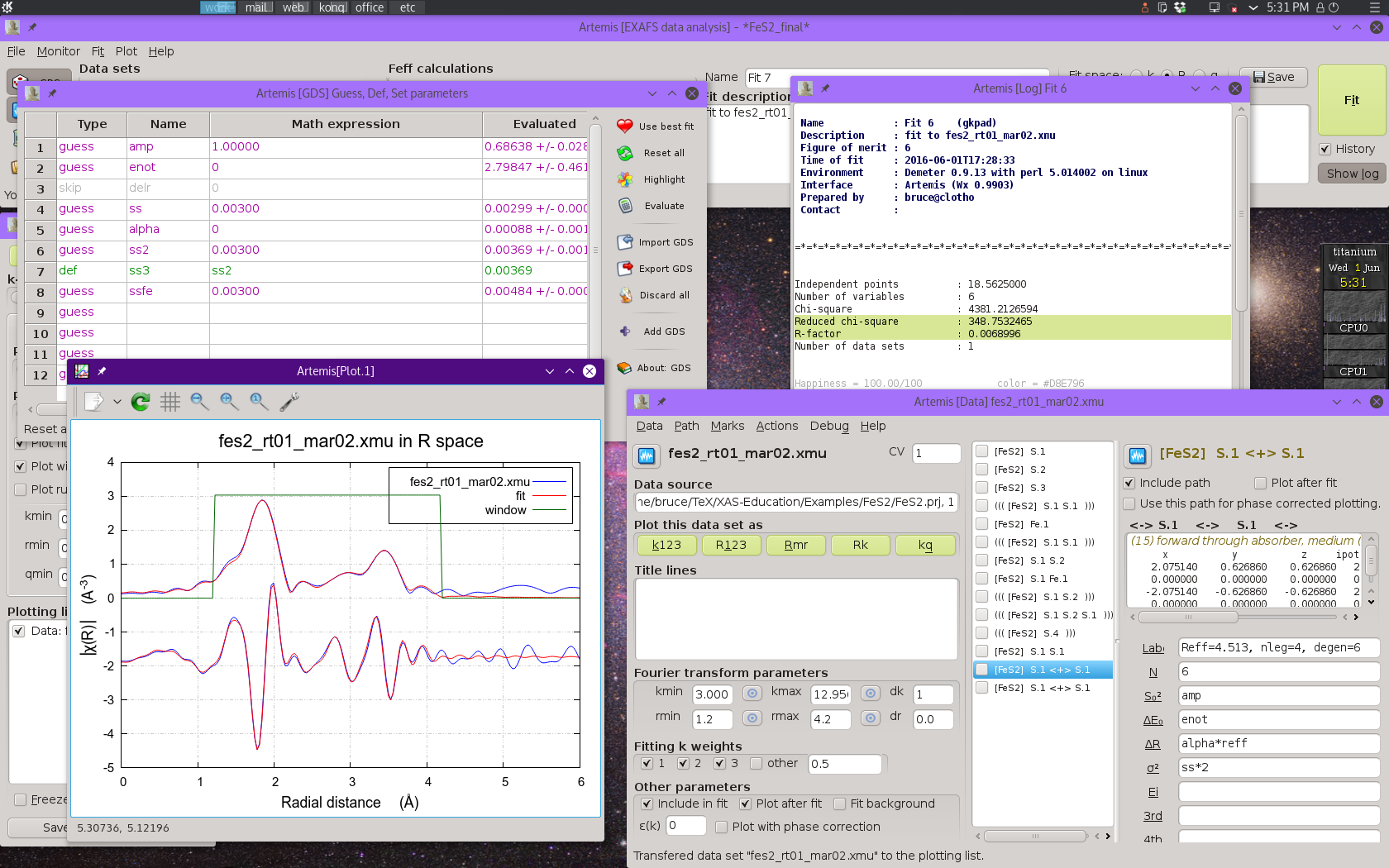
Fig. 13.14 Result of the final fitting model including four single scattering paths and five multiple scattering paths.
These three collinear multiple scattering paths are introduced into
the fit without needing to introduce new guess parameters.
The alpha*reff parameterization for ΔR works just as well
for these paths. Their σ2 parameters are expressed in
terms of the first shell σ2 following the example of
Hudson, et al.
- E. A. Hudson, P. G. Allen, L. J. Terminello, M. A. Denecke, and T. Reich. Polarized x-ray-absorption spectroscopy of the uranyl ion: comparison of experiment and theory. Phys. Rev. B, 54:156–165, Jul 1996. doi:10.1103/PhysRevB.54.156.
Please review the lecture notes for this fitting example for discussions of all the decisions that went into creating this successful fitting model.
DEMETER is copyright © 2009-2016 Bruce Ravel – This document is copyright © 2016 Bruce Ravel
This document is licensed under The Creative Commons Attribution-ShareAlike License.
If DEMETER and this document are useful to you, please consider supporting The Creative Commons.