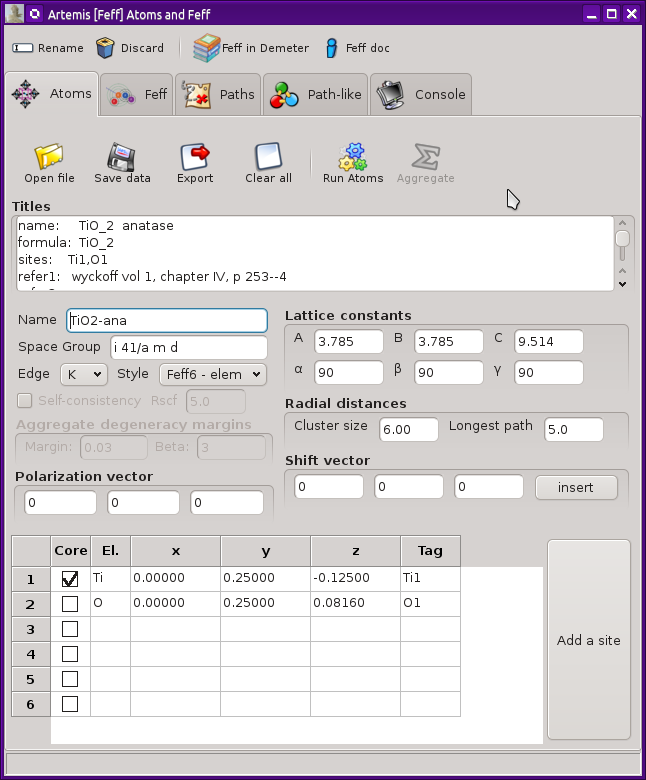
The Atoms and Feff Window
When you import crystal data from an atoms.inp or CIF file, three things happen:
A new ATOMS and FEFF window is created for interacting with
the structural data and
the various controls are set to values taken from the
atoms.inp or CIF file,
A message is written to the status bar in the Main window.
An entry is placed in the FEFF list on the main window.
You can also import a feff.inp file
directly. This is discussed in the
next section
This new window looks like this. In this example, crystal data for
anatase TiO2 have been imported from an
atoms.inp file.
At the top of the window is a tool bar with four buttons. The first
of these is used to change the name of this FEFF calculation.
Among other things, this is the label used in the FEFF list on the
Main window. The second button is used to discard the FEFF
calculation and this window. The final two buttons open a web
browser and take you either to the FEFF document or to this page
in the ARTEMIS document.
There are a series of tabs across the top. These will contain
different stages of the structural calculation. Here we will examine
the ATOMS tab. The other tabs will be examined in the following
sections.
Crystal data
The crystal cell data – including lattice constants and angles, the
space group symbol, and the elements of the shift vector – are placed in
text boxes for easy editing. The coordinates of the unique sites are
listed in the grid at the bottom of the window. The absorber is
chosen by clicking one of the boxes on the
Core column.
Remember that FEFF considers numbers with 5 digits of precision
after the decimal point. 0.333 is not the
same thing as 0.33333. You may, however,
enter things like 1/3 and avoid the
precision issue entirely.
As a new feature compared to earlier versions of ATOMS, there are
two radial distances. The cluster size determines the extent of the
cluster expanded into the feff.inp. This
should usually be set to something rather large, 9 Å is often a
good default. This probably (but not always!) assures that the
cluster in the feff.inp file is adequately
large to include all unique potentials and has all atom types
sufficiently well bounded that the muffin tin potentials are likely to
be be computed reasonably well.
The second distance will set the value of RMAX in the feff.inp file. In
general, you do not want this to be much larger than the extent of the
data you intend to analyze. 5 Å or 6 Å is usually the
largest sensible value for longest path. The reason for this is that
the pathfinder part of FEFF has been rewritten for this version
of ARTEMIS. While the new pathfinder implementation offers a
number of useful new features, it is substantially slower than
FEFF's native pathfinder. In any case, there is no
benefit to computing paths that you will never use in your fit.
The absorption edge for the calculation is chosen from the menu to the
left of the lattice constant area. This is usually determined from
the input data, but may need to be explicitly selected. If not
specified in the atoms.inp file, the edge
will be set to K for element lighter than Ce (Z=58), and to LIII
for heavier elements.
The style menu is another new feature in this version of Atoms. It is
used to set how the list of unique potentials is determined from the
elements in the atoms list. The choices are
-
elements
-
Each unique element species is assigned a potential number.
-
tags
-
Each unique tag is assigned a potential number.
-
sites
-
Each crystallographic site is assigned a potential number.
Remember that FEFF only allows for 7 unique potentials other than
the absorber. The tags and sites options can often result in more
than 7 potnatials, which will result in an unrunnable
feff.inp file. Specifying unique
potentials by tags is a good way of differentiating between dissimilar
atoms of the same species. For example, in an oxygenyl species, it is
often useful to give the axial oxygen atoms a different potential from
the remaining oxygens by using the tags option.
Here is an example of an atoms.inp file
for sodium uranyl acetate, which contains two very short axial oxygen
atoms double bonded to the uranium atoms at about 1.8 Å and a
number of equatorial oxygen atoms at a much longer distance. The
axial and equatorial oxygen positions are distinguished by their tags
and will given separate unique potentials when using the tags style.
The assignment of potential indeces is explained in detail and with
examples in the extended explanations chapter.
title Templeton et al.
title Redetermination and Absolute configuration of Sodium Uranyl(VI) triacetate.
title Acta Cryst 1985 C41 1439-1441
space = P 21 3
a = 10.6890 b = 10.6890 c = 10.6890
alpha = 90.0 beta = 90.0 gamma = 90.0
core = U edge = L3
atoms
! elem x y z tag occ
U 0.42940 0.42940 0.42940 U 1.00000
Na 0.82860 0.82860 0.82860 Na 1.00000
O 0.33430 0.33430 0.33430 Oax 1.00000
O 0.52420 0.52420 0.52420 Oax 1.00000
O 0.38340 0.29450 0.61100 Oeq 1.00000
O 0.54640 0.24430 0.50070 Oeq 1.00000
C 0.47860 0.22600 0.59500 C 1.00000
C 0.50880 0.12400 0.68620 C 1.00000
Polarization
A FEFF calculation considering linear polarization can be
triggered by setting one or more non-zero values for the polarization
vector.
This vector sets the value of the POLARIZATION
keyword in the resulting feff.inp. The
value written in the feff.inp file is the
value that will be used in the pathfinder and when
computing the path contributions. That is, if you edit the
POLARIZATION in the
feff.inp file, the edited value will take
precedence over the value specified here.
 FEFF's ELLIPTICITY keyword is not supported at this time.
That means the trick of modeling
“polarization in the plane” is not yet
supported by ARTEMIS.
FEFF's ELLIPTICITY keyword is not supported at this time.
That means the trick of modeling
“polarization in the plane” is not yet
supported by ARTEMIS.
Atoms tool bar
The toolbar across the top of the ATOMS tab offers several functions.
Clicking the open button will post the standard file selection dialog
for importing a new atoms.inp or CIF file.
This is more useful in the stand-along version of ATOMS than in
ARTEMIS where the crystal data file imported in other ways.
Right clicking this button will post the
recent files dialog populated with recently imported
atoms.inp,
feff.inp, and CIF files.
 The save button will prompt you for a filename for an output
atoms.inp saving the current state of the
tab.
The save button will prompt you for a filename for an output
atoms.inp saving the current state of the
tab.
Clicking the export button will post the dialog on the right, which
offers several different kinds of output files based on the crystal
data.
The “Feff6” and
“Feff8” options will write input files for
FEFF6 and FEFF8.
The “Atoms” option write the same file as
the save button.
The “P1”
option writes the crystal data to an atoms.inp
file using the P 1 space group and with a
fully decorated unit cell. The “Spacegroup”
option writes a file that fully describes
the space group.
The “Absorption” option
writes a file containing some calculations based on tables of X-ray
absorption coefficients.
“XYZ” and
“Alchemy” are formats that are commonly
understood by molecule rendoring software.
“Overfull”
is an “XYZ” file with the contents of
the unit cell in Cartesian coordinates and with all
atoms near a cell wall replicated near the opposite cell wall. The
purpose of this output type is generate nice figures of unit cells
with decorations on all the corners, sides, and edges.
The clear button is used to clear all data from all controls on the
ATOMS tab.
The run button is pressed to convert the crystal data
into input data for FEFF. Pressing this displays
the next tab.
The aggregate button is discussed in detail in
a later section
|