4. The Atoms and Feff Window¶
When you import crystal data from an atoms.inp or CIF file,
three things happen:
- A new ATOMS and FEFF window is created for interacting with the structural data and the various controls are set to values taken from the atoms.inp or CIF file,
- A message is written to the status bar in the Main window.
- An entry is placed in the FEFF list on the main window.
You can also import a feff.inp file directly. This is discussed in the
next section
This new window looks like this. In this example, crystal data for
anatase TiO2 have been imported from an atoms.inp file.
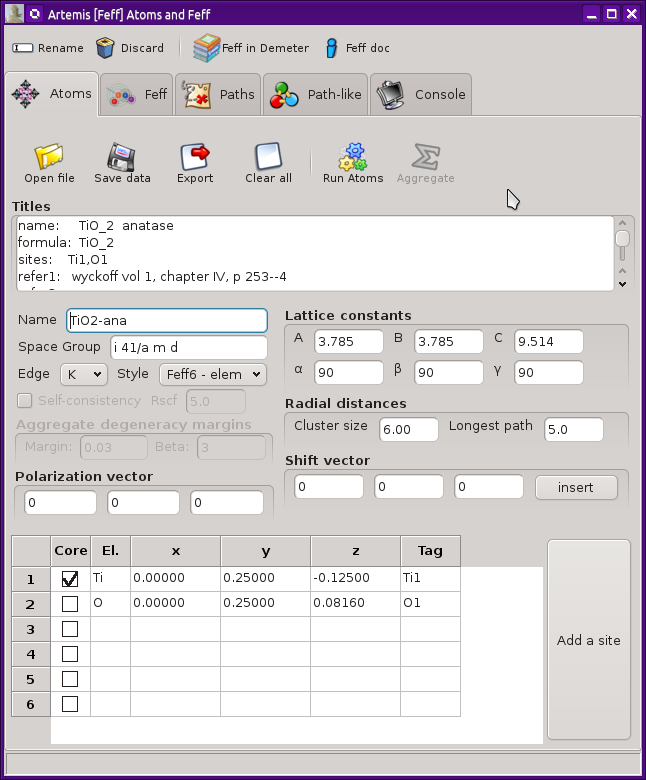
Fig. 4.1 The Atoms and Feff window.
At the top of the window is a tool bar with four buttons. The first of these is used to change the name of this FEFF calculation. Among other things, this is the label used in the FEFF list on the Main window. The second button is used to discard the FEFF calculation and this window. The final two buttons open a web browser and take you either to the FEFF document or to this page in the ARTEMIS document.
There are a series of tabs across the top. These will contain different stages of the structural calculation. Here we will examine the ATOMS tab. The other tabs will be examined in the following sections.
The toolbar across the top of the ATOMS tab offers several functions.
Clicking the open button will post the standard file selection dialog
for importing a new atoms.inp or CIF file. This is more useful in the
stand-along version of ATOMS than in ARTEMIS
where the crystal data file imported in other ways.
 Right clicking this button will post the
recent files dialog populated with recently imported
Right clicking this button will post the
recent files dialog populated with recently imported
atoms.inp, feff.inp, and CIF files.
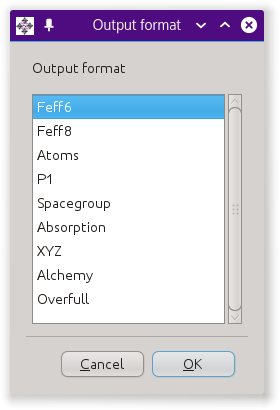
Fig. 4.2 The save button will prompt you for a filename for an output
atoms.inp saving the current state of the tab.
Clicking the Export button will post the dialog in Fig. 4.2, which offers several different kinds of output files based on the crystal data.
- The Feff6 and Feff8 options will write input files for FEFF6 and FEFF8.
- The Atoms option write the same file as the save button.
- The P1 option writes the crystal data to an
atoms.inpfile using theP 1space group and with a fully decorated unit cell. - The Spacegroup option writes a file that fully describes the space group.
- The Absorption option writes a file containing some calculations based on tables of X-ray absorption coefficients.
- XYZ and Alchemy are formats that are commonly understood by molecule rendering software.
- Overfull is an “XYZ” file with the contents of the unit cell in Cartesian coordinates and with all atoms near a cell wall replicated near the opposite cell wall. The purpose of this output type is generate nice figures of unit cells with decorations on all the corners, sides, and edges, like Fig. 15.37.
The Clear button is used to clear all data from all controls on the ATOMS tab.
The Run button is pressed to convert the crystal data into input data for FEFF then displays the next tab.
The Aggregate button is discussed in detail in a later section
DEMETER is copyright © 2009-2016 Bruce Ravel – This document is copyright © 2016 Bruce Ravel
This document is licensed under The Creative Commons Attribution-ShareAlike License.
If DEMETER and this document are useful to you, please consider supporting The Creative Commons.